
| 異染性腦白質退化症 (Metachromatic leukodystrophy) | |
|---|---|
 | |
| 腦硫脂的結構式 | |
| 症状 | 失智症 |
| 类型 | sphingolipidosis[*], rare hereditary metabolic disease with peripheral neuropathy[*], rare genetic epilepsy[*], unclassified primitive or secondary maculopathy[*], metabolic disease with dementia[*], hereditary retinal dystrophy[*], rare dyslipidemia[*], neurometabolic disease[*], sphingolipidosis with epilepsy[*], sulfatidosis[*], disease of a particular individual[*] |
| 分类和外部资源 | |
| 醫學專科 | 內分泌學、神經學 |
| ICD-11 | 5C56.02 |
| ICD-10 | E75.2 |
| ICD-9-CM | 330.0 |
| OMIM | 250100 |
| DiseasesDB | 8080 |
| MedlinePlus | 001205 |
| eMedicine | ped/2893 |
| MeSH | D007966 |
| Orphanet | 512 |
異染性腦白質退化症(Meta-chromatic leuko-dystrophy、MLD,亦稱:異染性腦白質營養不良症、芳基硫酸酯酶A缺乏症<Arylsulfatase A deficiency/ARSA deficiency>、Greenfield's disease)為一種溶酶體貯積症(lysosomal storage disease),將MLD的神經纖維切片以結晶紫染色結果不呈紫色而呈黃褐色、故名之"異染性"(meta-chromatic),一般歸於腦白質營養不良(leukodystrophies)的類別、同於鞘脂質代謝障礙(sphingolipidoses),因其影響神經鞘脂質(sphingolipids)的代謝。而腦白質營養不良影響髓磷脂的"生長"和/或"發展",而"脂質"的被覆是作為周圍神經纖維的"阻隔體"(insulator)貫穿整個中樞和外週的神经系统。MLD亦涉及脑苷脂的積累。 異染性腦白質退化症、如同大多數的"酶缺陷"(enzyme deficiency)一樣,存有「常染色體隱性遺傳模式」。
原因
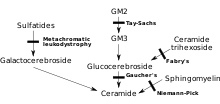
MLD是因染色體#22q13.31ARSA基因之缺失,直接導致芳基硫酸酯酶A(ARSA)的缺乏所引起, 並且通常其特徵在於"酶的活性"不足人體所能控制的10%。 如果沒有這種酶,腦硫脂在身體的許多組織中積聚無法分解(MLD 有較高水平的腦硫脂;相對的、阿茲海默症有較低水平的腦硫脂),最終會破壞神經系統的"髓鞘"(myelin sheath)。髓鞘是一種脂肪覆蓋體,作用是保護神經纖維。沒有髓鞘,在大腦的中樞神經(中樞神經系統-CNS)及外週神經(外週神經系統-PNS)的控制功能,其中包括肌肉的移動等活動,就"不再正常地運作"。最近的一項研究主張"腦硫脂"不完全對MLD的症狀負責,因為腦硫脂是無毒的。有人已提出"溶血腦硫脂"(溶血硫腦苷脂/lyso-sulfatide)由於其體外細胞毒性的作用而引起MLD。而"腦硫脂"已去除它的酰基、因之不完全會激起MLD病症。
遺傳學

MLD為一種「常染色體隱性遺傳模式」。在"每個出生嬰兒"的繼承概率如下:
- 如果父母雙方都是"帶原者":
- 25%(1/4)的小孩將會有此病症。
- 50%(2/4)的小孩將會成為帶原者(不過不會受到感染)。
- 25%(1/4)的小孩將不會有MLD -- 未感染的小孩不會成為帶原者。
- 如果父母只有一方"感染",而另一方"沒有感染MLD":
- 0%(0)的小孩將會有此症狀 -- 父母只有一方受到感染,另一方總是遺傳正常基因。
- 100%(4/4)小孩將會成為帶原者(不過不會受到感染)。
- 如果父母只有一方為帶原者,而另一方沒有感染MLD:
- 50%(2/4)的小孩將會成為帶原者(不過不會受到感染)。
- 50%(2/4)的小孩將不會有MLD -- 未感染的小孩不會成為帶原者。
除了這些遺傳模式出現繼承概率外、還存有一種「偽-ARSA缺乏症(pseudo-(Arylsulfatase A) deficiency)」,影響到7%-15%的人口。 有"偽-缺乏症"的人沒有任何MLD的問題,除非他們也受到感染。以目前的診斷測試分析、"偽-缺乏症"呈現出"低酶"的水平,但"腦硫脂"的運作正常、因此MLD症狀不存在。這種"偽-現象"造成"傳統的檢查法"至"新生兒篩查法"(Newborn Screening)嚴重的誤判,因此"更新的篩選法"也正在研發。
欲了解更多信息,請參見隱性基因和顯性關係等條目。此外,也可諮詢<MLD基金會>的MLD遺傳學網頁。
發病率
估計"異染性腦白質退化症"在全世界人口中的发病率分佈在<1比40,000>至<1比160,000>之間。 有更高的發病率發生在"特定的遺傳隔離群體"(certain genetically isolated populations)裡、如<1比75>的以色列哈巴尼猶太人(Habbani Jews、從阿拉伯南部移民到以色列的一小群猶太人)、<1比2,500>在美國纳瓦霍族保留地的西部地區,以及<1比8000>在以色列的阿拉伯群體裡。
症狀和MLD形式
如同其他許多會影響"脂質代謝"(lipid metabolism)的遺傳性疾病一樣,MLD也有多種病發期的MLD形式,如嬰幼兒後期、青少年儿童期,及成人期。
- 在"嬰幼兒後期"的MLD形式,這是MLD的最常見的病發期(佔病症人口的50-60%之間),受感染的嬰幼兒在出生第一年後行走困難,病發期通常分佈在出生後15-24個月之間。 發病症狀包括肌肉萎縮及無力、肌肉強直、發育遲緩、視力逐漸喪失、導致失明、抽搐、吞嚥障礙、瘫痪和癡呆。孩子可能會轉為昏迷。未及時治療,多數患有這種形式MLD的孩子會死於5歲左右,通常會更快。
- 青少年兒童期的MLD形式(發病於3至10歲之間),通常開始於學校的表現落後、智力減退、癡呆、或一些精神症狀出現,病發的症狀相似於"嬰幼兒後期",但病情發展較慢。死亡年齡為"變量"(variable),但通常介於症狀發作期的10歲至15歲之間,然而有些青少年可以繼續存活至發病後數十年或更長的時間。
安寧療法、或者針灸療法可以對MLD症狀有許多幫助,通常可以改善生活品質和延長壽命。
帶原者和他們的家人相比之下呈現"低酶水平"("正常的酶水平"因家庭而異),不過即使是低酶水平仍足以處理身體的"腦硫脂積累"現象。
治療方法
由於神經系統髓鞘的缺損在時下的醫學治療上屬於不可逆性,故而目前還沒有確切的治療或治愈MLD的方法,僅能針對MLD病狀做改善、或安寧性治療。"較長的青少年"或"成人"開始發病及"嬰幼兒後期"的患者在症狀顯示後接受治療,會拘限於疼痛和病症處理等之問題;有少數患者可用Vighafvine(抗抽筋藥)來緩解痙攣的疼痛。在症狀前期之"嬰幼兒後期"的MLD患者,以及那些青少年或成年MLD患者,要麼出現"症狀前期病徵"或者顯示"輕度至中度的症狀";而對於可選擇骨髓移植(包括幹細胞移植)的患者,目前正在研究,看它是否可能讓MLD的症狀進展放緩、或停止MLD在中樞神經系統中的症狀進展。然而,結果顯示MLD的症狀進展在周圍神經系統一直沒有顯著的放緩,而這些長期治療的效果好壞參半。
幾個未來的治療方案,目前正在進行研究。這些措施包括基因治療、酶替代療法(Enzyme replacement therapy/ERT)、底物減少療法(Substrate reduction therapy/SRT),及"潛在性酶增強療法"(potentially enzyme enhancement therapy/EET)等。
一組國際研究人員及基金會在2008年組成國際MLD註冊單位 (页面存档备份,存于互联网档案馆)創建和管理知識共享資源庫,包括MLD的自然史(NHS)基礎。該組織包括科學,學術和工業資源。然而、之後註冊單位從來沒有開始運作。
臨床試驗
骨髓和幹細胞移植療法
基因治療
目前正在研究兩種不同的基因療法、以之來治療MLD病症。
-
基因治療與自體(Autotransplantation)幹細胞移植 -- 意大利的研究人員在"聖拉斐爾特勒松"研究院 (页面存档备份,存于互联网档案馆)(San Raffaele Telethon Institute)測試"結合基因治療幹細胞移植"的新方法。 計劃為招募MLD患者進行"第I/II期"的臨床試驗、時程自意大利當局2010年3月24日批准後正式啟動。最初招募的試驗組為8名MLD患者,計劃完成於2013年三月中旬。該試驗是測試"自體"(Autologous blood donation、利用患者自身的細胞)的造血幹細胞移植(HSCT)機制、在經由"血細胞路徑"(route of the blood cells)將"基因修飾"(genetic modification)過再傳送<"超治療"(強調性質)的"芳基硫酸酯酶A"(ARSA enzyme)>到達"神經系統"後之有效性和安全性的評測。即利用患者自身的"幹細胞"(stem cell)與基因來修正,應該可以減少或消除"嫁接"(graft)對於"宿主"(host)的併發症。並且能夠對於出現在MLD病患身體裡的ARSA提供長期的解決方案。在"標準試驗"(bench)及動物試驗下均顯示出正向成果。研究人員在2013年7月公佈二年來第一批3名患者的療程成效。結果被認為有研發潛力。
- 入選標準為呈現"前期症狀"之"嬰幼兒後期"的小孩,及呈現"前期"和"早期"症狀的青少年。查看入選標準及試驗協定在這兒。
- 該試驗是在意大利米蘭的單一中心進行。所有費用都由研究人員支付。這是一項為期3年的研究。在2013年3月,八名初級試驗患者的最後一人開始接受治療。而且一項關懷體恤的探訪活動正在進行。(2013年3月)
- 亞洲自體幹細胞基因移植 -- 亞洲首宗由香港大學李嘉誠醫學院、大陸深圳市第二人民醫院,及台灣的臺大醫院。於2014年9月為成人期MLD形式的後期患者俞潤潤,於深圳市第二人民醫院進行亞洲首宗自體幹細胞基因移植。 至今患者恢復情況良好。
-
腦基因治療 -- 在2013年的三月下旬於巴黎開始招募"第I/II期"臨床試驗病患、進行"腦基因"(Intracerebral Gene Therapy)治療的臨床試驗,其中特殊的"向量體"(vector)載送"基因修飾過的材料"直接注射到大腦中的十幾個地點。希望"修正過的細胞"來製造"酶"然後擴散到大腦的周邊地區。在實驗室的延伸工作及一些令人鼓舞的ALD研究 (页面存档备份,存于互联网档案馆),提供了這項試驗的依據。
- 招募5個年紀較大的小孩、年齡介於4歲及6歲之間、進行為期兩年的研究。
- 必須在過去的12個月裡第一次出現過症狀。
- 附加的試驗入選標準和資訊可以在這兒找到
酶替代療法(ERT)
-
希雷修曼(Shire Human)基因療法 (页面存档备份,存于互联网档案馆)(希雷HGT)為英國希雷PLC (页面存档备份,存于互联网档案馆)的一個部門,2012年1月開始為它們的鞘内注射(脊髓腔內注射/IT)"HGT-1110ERT產品"之臨床試驗招募病患。
- 入選標準
- 第一次症狀出現在2歲半之前、而目前為7歲或年齡更小的幼兒。
- 在門診走廊 -- 只用一隻手扶著能夠走10步。
- 額外的臨床試驗資訊和入選標準,可以在MLD基金會網站這兒,及在臨床Trials.gov (页面存档备份,存于互联网档案馆)網站上找到。
- 這次臨床試驗為期38週、18個兒童以三種不同的劑量分組,且分散在多個地點進行。"不治療"(no treatment)安慰劑組從2012年6月的試驗裡刪除。
- 患者必須到五個試驗地點之一進行每間隔一周的酶注射:丹麥哥本哈根,法國巴黎,德國蒂宾根,澳大利亞悉尼,或巴西阿雷格里港。而阿根廷的"德爾基"(Derqui)正等待批复。
- 2013年12月被批准開始從一個新的供應商獲得一個新的"鞘内注射埠口"(intrathecal port)。更詳細資訊請參見MLD基金會網頁。
- HGT-1110ERT在歐洲和美國均是"孤兒產品"(orphan product)狀態。
- 歷史: 希雷在2010年暫停"元酶靜脈ERT"(Metazyme intravenous ERT)產品的開發。在2008年從賽門艾克(Zymenex)(由"希雷"收購後更名為HGT-1111)收購時它只是一個臨床試驗,在歐洲的"第I/II期"臨床試驗之後它被證明不具有足夠的功效。最初的研究2008年9月完成,而且伴隨著"停止供應產品給試驗參與者"的擴展研究於2010年10月完成。
- 入選標準
底物減少療法(SRT)
- "白莫林南方公司"(Biomarin South、於2013年1月被"白莫林"收購之前屬於拉凱隆(Zacharon)公司)在美國聖地亞哥已經啟動了"溶酶體貯積症"(Lysosomal storage disorders/LSDS)的藥物研發計劃。這個計劃是基於使用檢測來量測"腦硫脂積累"(sulfatide accumulation)程度,也是為藉由"培養過的纖維細胞"(cultured fibroblast)來作為研究和開發MLD"小分子藥物"(small molecule drug)的方法。(此方法不同於其他的方法像是由所測得的酶活性來研發有效性藥物) 至2011年7月、拉凱隆(Zacharon)公司已先開始進行化驗分析工作,主要是為了"溶酶體貯積症"而進行藥品研發,如此他們可以被聘僱來研究和開發MLD的藥物。(2013年3月)
- "庫珀健康系統"(Cooper Health System、新澤西州)在2009年贊助治療MLD之臨床試驗,以確定"維生素K拮抗劑"(华法林)的安全性和有效性。眾所周知所發表的報告顯示SRT是沒有什麼成果。(2013年3月)
自然史研究
按此了解更多資料(目前為2014年2月)
由MLD基金會提供研究和臨床試驗的更新資訊
引用
来源
- 本文的某些部分出自美國神經學疾病與中風研究所(National Institute of Neurological Disorders and Stroke)的公共領域文章:
- NINDS Metachromatic Leukodystrophy Information Page. [2009-06-07]. (原始内容存档于2009-06-03).
參見
- 髓鞘再生研究計劃(The Myelin Project)
- 羅倫佐的油
- 髓磷脂修復基金會 (页面存档备份,存于互联网档案馆)
- 嚴重免疫缺陷綜合症(Severe combined immunodeficiency/SCID)
- 氯苯甲嗪(Meclizine/HCL/Meclizine 25mg Tab/美克洛嗪、美克旅鎮片、敏可靜、美克利靜、保暈錠/暈眩止吐用藥/白色圓扁形/錠劑)
- 鞘内注射泵(Intrathecal pump)
- 多硫酸酯酶缺乏症(Multiple sulfatase deficiency)
- 曲吡那敏(Tripelennamine)
- 硫酸酯酶(Sulfatase)
- 22q13缺失綜合症(22q13 deletion syndrome)
- 腦苷−硫酸酯酶(cerebroside-sulfatase)
- PNPLA3(PNPLA3)
- 鞘磷脂磷酸二酯酶1(Sphingomyelin phosphodiesterase 1)
- 腎上腺腦白質失養症(ALD)
- 臺灣腦組織資源聯盟建置策略(臺灣腦庫)
外部链接
- 全球性MLD专门組織
- 異染性腦白質營養不良及溶酶體病組織
- Australian Leukodystrophy Support Group (Australia)
- Bethany's Hope (Canada) (页面存档备份,存于互联网档案馆)
- ELA, The European Leukodystrophy Association (页面存档备份,存于互联网档案馆)
- The Evanosky Foundation (页面存档备份,存于互联网档案馆)
- Hide & Seek Foundation for Lysosomal Disease Research (页面存档备份,存于互联网档案馆)
- New Zealand Organisation for Rare Disorders (页面存档备份,存于互联网档案馆)
- The Stennis Foundation
- 有關MLD的進一步信息、治療、遺傳學,和當前的研究項目
- Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy (页面存档备份,存于互联网档案馆)
- The Evanosky Foundation
- MLD Foundation
- The Stennis Foundation (页面存档备份,存于互联网档案馆)
- 其它參考鏈接
- 財團法人罕見疾病基金會(Taiwan Foundation for Rare Disorders/TFRD) (页面存档备份,存于互联网档案馆)
- Biomarin (页面存档备份,存于互联网档案馆)
- Shire HGT (页面存档备份,存于互联网档案馆) drug development pipeline
- 2008 eMedicine article (页面存档备份,存于互联网档案馆) about MLD by Ikeda & Moore of UCLA and Steiner of OHSU
- NIH/GeneReviews overview of MLD written by Arvan Fluharty of UCLA (updated August 2011)
- 台灣神經罕見疾病學會(Society for Neurological Rare Disorders-Taiwan - About TBTRC) (页面存档备份,存于互联网档案馆)
- 臺灣腦組織資源聯盟建置策略(臺灣腦庫) – 國家衛生研究院論壇 (页面存档备份,存于互联网档案馆)
| ||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||