
 | |
| 臨床資料 | |
|---|---|
| 商品名 | 百悦泽 Brukinsa |
| 其他名稱 | BGB-3111 |
| AHFS/Drugs.com | Monograph |
| 核准狀況 | |
| 给药途径 | 口服给药 |
| 藥物類別 | 布魯頓氏酪氨酸激酶 (BTK) 抑制劑 |
| 法律規範狀態 | |
| 法律規範 | |
| 识别信息 | |
| CAS号 | 1691249-45-2 |
| PubChem CID | |
| PubChem SID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| 化学信息 | |
| 化学式 | C27H29N5O3 |
| 摩尔质量 | 471.5509 |
| 3D模型(JSmol) | |
| |
| |
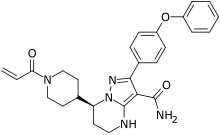
泽布替尼(英語:Zanubrutinib,商品名:百悦泽,英語:Brukinsa)是一種治療被套細胞淋巴瘤的藥物,它被定義為一種布魯頓氏酪氨酸激酶(BTK)抑制劑,並被製作成膠囊,患者通過口服方式服用此藥品。
該藥品由百濟神州研發,2012年6月開始立項,2019年11月在美国上市,成為首個由中华人民共和国研發的在美國獲批上市的抗癌新药。2020年6月3日,該藥獲准在中国大陆上市,成為該國首個上市的由該國生產的BTK抑制劑。
開發過程
百濟神州開發團隊要先尋找可以模仿 ATP adenine 的雜環,不過,過去已經有諸多文獻探討 BTK 抑制劑的構效關係,開發團隊不必從零開始尋找,就以已經上市的 Ibrutinib 開始。然開發團隊非直接拿 Ibrutinib 的 pyrazoloadenine 做研究,反而用分子內氫鍵手法修改 pyrazoloadenine 基團(下圖中的 series I),設計出的新化合物,新化合物會在分子內形成氫鍵,模仿 pyrazoloadenine 基團

有文獻指出,使用分子內氫鍵 (intramolecular hydrogen bond, IMHB) 手法模仿雜環結構可以提高藥物水溶解度和穿過細胞膜能力,改善藥動特性,亦可提高藥物效價(低濃度就可以有強烈藥理反應)。在藥物設計時,為了提高藥物對細胞膜穿透力(藥物才能進入細胞發揮作用),便需要提高藥物親脂性,也就是降低藥物水溶解度,可能使藥物難以做成製劑(舉凡針劑等需要溶解於水中),或(如果做成錠劑或膠囊)難以在消化道溶解,阻礙藥物吸收(錠劑在腸胃道裡要先崩散成顆粒,再溶解成小分子,方能被吸收,因此藥物不能太水溶性,也不能太脂溶性),當碰到如此窘境時,可以用 IMHB 手法修飾藥物。以 IMHB 修飾的化合物,會存在兩種構型(見下圖中 B),分別是無分子內氫鍵 的 “open” 構型(見圖十二 B 左)和有分子內氫鍵的 “close” 構型(見下圖中 B 右),兩種構型會達成一個平衡。當化合物處於極性環境時(見下圖中 A),環境中的極性分子(如水)與化合物之間形成氫鍵 ,讓 “open” 與 “close” 的平衡偏向 “open”,也因為化合物與水形成氫鍵,讓化合物得以溶於水中;相反的,如果化合物處於非極性(見下圖中 C),環境中沒有分子可以與化合物形成氫鍵,因而自己在分子內形成氫鍵,平衡偏向 “close” 構型,使得整個化合物變得偏向親脂性(因為極性基團被遮蔽)。IMHB 讓藥物能在不同環境下透過構型改變自己的極性,當在需要溶解於水中時,藥物變成親水性,得以溶於水裡;當藥物要穿過親脂性的細胞膜時,藥物變成親脂性,得以穿過細胞膜發揮藥效。文獻亦指出,透過 IMHB 修改雜環,因為分子內氫鍵使藥物構型也能與雜環一樣平坦,不影響藥物與蛋白質表面結合。

開發團隊合成一系列化合物,結構類似上上圖中 series I,並作藥理活性評估,這些化合物對 BTK 的效價不錯(下圖的化合物 I-2),確認 IMHB 在這藥物上是可行的
| 化合物 I-2 | 化合物 I-3 |

|

|
接著開發團隊合成三環化合物(series II),並作藥理實驗。發現 series II 具有高 BKT 效價與專一性。一開始,團隊先合成三環化合物 5,其結構上沒有前述說很重要的藥效基團 acrylamino,所以它是可逆抑制劑,IC50 (抑制 50% 某個生化功能所需要的藥物濃度)17 nM。當把 acrylamino 接上化合物 5 形成化合物 6,IC50 降低到 0.32 nM(只需要比較少的劑量就能達到一半效果),然化合物 6 在老鼠實驗上,生體可用率低。團隊猜想低生體可用率可能是因為三個芳香環導致的高剛性(芳香環結構是一平面,因此芳香環在藥物設計上可以固定藥物在特定構型,以方便藥物嵌入蛋白質內)。猜測團隊的意思是芳香環太多使藥物水溶解度不好,影響藥物生體可利用率,所以解決辦法就是減少芳香環數量,以其他環取代芳香環。
| 化合物 5 | 化合物 6 | 化合物 7 | 化合物 8 | 化合物 9 | 化合物 10 | 化合物 11 | 化合物 12 |
 |
 |
 |
 |
 |
 |
 |
 |
因此團隊把其中一個芳香環以脂族環,合成出化合物 8, 9, 10, 11, 和 12。這五個化合物的效價都與化合物 6 相當,且都可以抑制 BTK pY223 (在第 223 位上磷酸化的 BTK,團隊要確認藥物也能抑制已經活化的 BTK;未活化和已活化的蛋白質,在結構上有些變化,要確認即便結構起了點變化,藥物依然能發揮作用)。可惜的是,化合物 5-12,除了化合物 6(選擇性七倍),其他化合物對於 EGFR 沒有選擇性(意即藥物不只會抑制 BKT,還會抑制 EGFR)(EGFR IC50/BKT IC50,數字越大,選擇性越好)。除了選擇性問題外,低藥動特性的問題並沒有因為把芳香環換成其他脂族環而有所提高,以化合物 8 和 11 為例,在老鼠實驗上脾細胞佔率分別是 22% 和 44%(團隊實驗是給老鼠服用每公斤10 毫克劑量,四個小時後看脾臟細胞裡的藥量;猜測是因為脾臟充滿淋巴細胞,因此取脾臟細胞,再加入 BKT 看脾臟內有多少化合物-BKT 複合物,判斷有多少藥在體內)。
團隊認為低藥動特性的原因是三環所致高剛性,所以團隊合成雙環來看問題能不能被解決(前述的雙環三環是指兩個或三個還連在一起。團隊合成以 imidazopyrazole 為主結構的化合物13-16, 化合物 13 和16的 IC50 不錯(分別是2.7和 0.13 nM)。其中化合物 13 的選擇性最高(77 倍),但效價不如化合物 16,且化合物 16 藥動特性仍不理想(脾細胞佔率 56%)。
| 化合物 13 | 化合物 14 | 化合物 15 | 化合物 16 |
 |
 |
 |
 |
為解決化合物 16 差藥動特性,團隊改以 pyrazolopyrimidine 作為雜環合成出化合物 17, IC50 變為 860 nM。團隊為了讓化合物少一點芳香環的結構與利用雙環達到共平面,將化合物 17 還原成化合物 18,IC50 降低到 2.6 nM,進步了將近 300 倍。化合物 18 有兩個鏡像異構物,將異構物分開後 分別作藥理活性實驗,18a 的效價稍比18b 高,兩者選擇性都大於 60 倍。接著團隊把前述說的重要藥效基團(因為此基團帶有攻擊性,之後以 ”彈頭“ 稱之)移到苯環對位,合成出化合物 19,效價降低;彈頭移到苯環鄰位,合成出化合物 20,其效價比化合物 18 高出四倍。化合物 20 也有兩個鏡像異構物,20a 的效價比 20b 高達 340 倍之多,但 20a 的選擇性就沒有如化合物 18 和化合物 19 好。雖然化合物 18a, 18b 和 20a 在藥理活性上表現不錯,但在老鼠實驗上,生體可用率和口服吸收依然低下(20a口服生體可用率1.7%)。
| 化合物 17 | 化合物 18 | 化合物 19 | 化合物 20 | 化合物 21 |
 |
 |
 |
 |
 |
團隊以化合物 20 起始,將 phenoxyl 基團換成其他取代物, 試圖降低藥物脂溶性,以提高口生體可用率,合成出化合物 22-30。取代物包括比較大的基團(化合物 22-23)和比較小的基團(化合物 24-30)。以 cyclopropylmethoxyl (23a), methoxyl (25a) 或 chloride (29a) 取代 20a 的 phenoxyl,在藥理活性上並無太大改變;體外實驗發現這三個化合物溶解度和對肝臟微粒體穩定度都表現不錯(在 pH 7.4下,20a/23a/25a/29a 溶解度33.6/74.7/97.5/83.7 μM)。這三個化合物在老鼠身上,口服生體可用率也有進步(20a/23a/25a/29a:1.7/34.0/24.8/30.6%),藥動特性也表現優異(脾細胞81/77/82%)。但他們對 EGFR 的選擇性就沒如此好。

為了提高選擇性以及口服吸收,團隊以化合物 20 為始(猜測不用23a,25a或29a的原因可能是可能是修改 phenoxyl 沒辦法在口服吸收和選擇性都合宜,便修改另一個基團看看效果),把其中一個芳香環換成其他脂族環,合成出化合物 31-35。其中,消旋物 31, 32 效價與化合物 20 相當,化合物 34 和 35 的修飾讓效價降低 6 倍與 70 倍,表示 cyclopropyl 和 gem-methyl 基團太大,阻礙彈頭在適合位置攻擊 Cys481。把鏡像異構物分開,31a(即 Zanubrutinib), 32a, 33a 在藥理活性上表現優異,團隊以藥物讓酵素去活化的速率(kinact)/ 藥物抑制速率常數(KI) 評估藥物與蛋白質形成共價鍵的效果,結果顯示 31a ≈ Ibrutinib > 31b。接著,團隊拿 31a, 32a, 和 33a 與 Rec-1 (被套細胞瘤細胞)cells 做實驗,這三個化合物對於抑制細胞 BTK 表現 優異。團隊為避免 31a 的抗增生活性是因為它具有毒胞性(就像鹽酸可以殺死癌細胞,但不能作為抗癌藥),因此拿人類胚胎腎細胞 HEK293 和人類 B 淋巴球細胞瘤 細胞 Ramos cells (非 BTK 依賴的細胞)測試其存活率,實驗顯示 31a 對這兩細胞無毒殺作用。團隊再拿 31a 對瀰漫性大 B 細胞瘤細胞做實驗,其 IC50 (0.35 nM)與 ibrutinib 相當(0.30 nM)。31a 也對 EGFR 有很高的選擇性(337 倍)。

團隊將 31a 與 BTK 結合,並做 X 光晶體繞射看其與 BTK 的結合情況,並和 ibrutinib 做比較。顯然發現,這兩個化合物的結合姿態有點不同,與 ibrutinib 同都與 Glu475 (E475) 和 Met477 (M477) 形成氫鍵,但不同的是 31a 與 Met477 形成兩個氫鍵,比 ibrutinib 多一個。另外,也可以見在 Phe 450,藥物的芳香環與之形成 𝜋-𝜋 stacking,有個水分子與 Lys430 和藥物的 pyrazolyl 形成氫鍵。
最後,團隊拿 31a, 32a 和 33a 做老鼠實驗,發現 31a 相比其他兩個化合物,清除率相對低(31a/ 32a/33a:76.8/169/93.9 mL/min/kg)、血中最高濃度高(235/10.4/19.6 ng/mL),曲線下面積 AUC 高( 257/50.3/51.1 h・ng/mL),生體可用率高 (23.6/8.6/5.7 %)。因此拿 31a 做更進一步的實驗。
藥物合成

外部链接
- Zanubrutinib. Drug Information Portal. U.S. National Library of Medicine. [2020-06-05]. (原始内容存档于2020-12-06).
| ||||||||||||||||||||||||||||||||||||||||||||||||