
| 杜興氏肌肉營養不良症 | |
|---|---|
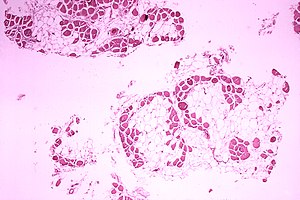 | |
| 杜興氏肌肉營養不良症患者的腓腸肌病理組織切片,其肌肉纖維被脂肪細胞所取代。 | |
| 症状 | 肌肉無力、站立困難、脊椎側彎 |
| 常見始發於 | 大約4歲 |
| 肇因 | 遺傳疾病(X染色體隱性遺傳) |
| 診斷方法 | 基因檢測 |
| 治療 | 物理治療、骨支架、外科手術、呼吸輔助器 |
| 预后 | 平均预期寿命約26歲 |
| 盛行率 | 5,000名男嬰中會有1名 |
| 分类和外部资源 | |
| 醫學專科 | 醫學遺傳學、小兒科 |
| ICD-11 | 8C70.1 |
| OMIM | 310200 |
| DiseasesDB | 3985 |
| MedlinePlus | 000705 |
| Patient UK | 杜興氏肌肉營養不良症 |
| Orphanet | 98896 |
杜興氏肌肉營養不良症(Duchenne Muscular Dystrophy,縮寫DMD)是一種相當嚴重的性聯遺傳肌肉失養症。男性病患大約在4歲開始就會產生肌肉無力的症狀,此後症狀即會開始快速惡化。通常最先從大腿即骨盆肌肉開始萎縮,之後則是上臂肌肉。本病會導致站立困難,患者大約在12歲之後就無法行走。受影響的肌肉會被脂肪組織佔據,因此看起來會較大塊。患者亦常見脊椎側彎或智能障礙。女性患者有時會表現些微症狀。
本病屬於性聯遺傳,約三分之二的患者疾病基因來自遺傳,三分之一則來自新突變。疾病相關基因位於X染色體上的DMD基因上,該基因負責了失養素的轉譯。失養素則與肌肉細胞膜的維持有關係。在孩子出生前可以進行基因檢測。患者血中的肌酸激酶值也會較高。
目前該病尚無有效的治療方法。物理治療、輔具,以及手術矯正都能幫助舒緩症狀。呼吸肌較弱的患者則可搭配呼吸輔助器。藥物則可使用皮質類固醇來減緩肌肉退化;抗抽搐藥物可以用於控制癲癇發作及肌肉不正常運動;免疫抑制劑則能延緩肌細胞的死亡及傷害。
約每5000名男嬰會有一人罹患此病,杜興氏肌肉營養不良症為最常見的肌肉失養症。患者平均預期壽命約為26歲,然而如果接受良好治療,患者亦可能存活至30到40歲。 此症由義大利那不勒斯的醫生Giovanni Semmola及Gaetano Conte分別於1834年及1836年提出報告,但症狀名稱是取自1861年在其書中詳細描述此病的法國神經學家Guillaume-Benjamin-Amand Duchenne。 另一種病情較輕的肌肉萎縮症稱為贝克型肌肉萎缩症(Becker's Muscular Dystrophy, BMD)是屬於DMD的亞型。
初期症狀
其他身體上的症狀有:
- 不穩步伐
- 容易跌倒
- 不良於行(跑步和跳)
- 愈來愈嚴重的行動困難
- 行路能力通常在12歲時消失
- 容易疲勞
- 輕度智能障礙(大概30%的患者)
- 骨骼發育畸形(有些個案為脊椎側彎)
- 肌肉發育畸形
- 小腿肌肉異常腫大(Pseudohypertrophy)(這些肌肉的正常組織被硬化組織所取代)
徵兆及檢驗方法
- 一般在5岁以前发病;
- 临床特点为进行性对称性肌无力,以肢体近端受累多见,起病常于下肢开始;
- 体查无肌颤,无感觉障碍,多伴有腓肠肌假性肥大;
- 肌电图呈肌源性损害;
- 肌活检表现为肌纤维长短不一,出现坏死与降解,纤维透明化,出现结缔组织与脂肪组织代偿增生,免疫组化分析可见dystrophin缺失;
- 有家族史,呈X连锁隐性遗传;
- 病情进行性加重。
治療
支持療法
- 提升自體體力
- 提升自體免疫力
- 心靈治療
晚期症狀
杜興氏肌肉營養不良症最終會影響到所有平滑肌,以及心臟和呼吸肌肉。患者很少能活过35岁。 患者一般由于呼吸衰竭或心功能紊乱而死亡。
療法研究
國際上曾經有人試圖用幹細胞移植的方法治療DMD,在老鼠身上獲得一定的進展。但用於人类患者身上尚沒有取得滿意的效果。目前基因治療仍在研究中,如何把DMD型改為BMD型,延長患者的壽命或針對“無義突變”發揮作用治療DMD/BMD,已經變成研究的熱點。
參考資料
外部連結
- 裘馨氏肌肉失養症(Duchenne dystrophy) (页面存档备份,存于互联网档案馆)
- 香港肌健協會 (页面存档备份,存于互联网档案馆)
- CDC's National Center on Birth Defects and Developmental Disabilities
- Muscular Dystrophy Campaign (UK) (页面存档备份,存于互联网档案馆)
- DMD Registry (页面存档备份,存于互联网档案馆)
- Parent Project UK
- Parent Project USA (页面存档备份,存于互联网档案馆)
- Duchenne/Becker Muscular Dystrophy, NCBDDD, CDC
- MUSCULAR DYSTROPHY Page on NCBI (页面存档备份,存于互联网档案馆)
- A support group for dads
- Report on U7 gene transfer studies PDF file。
- 中国神经肌肉疾病协会 (页面存档备份,存于互联网档案馆)
- 中華民國肌萎縮症病友協會 (页面存档备份,存于互联网档案馆)
- 中国DMD关爱协会
- 中国DMD关爱协会论坛
- 中国DMD肌营养不良注册登记 (页面存档备份,存于互联网档案馆)
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||