
| 克雅二氏病 | |
|---|---|
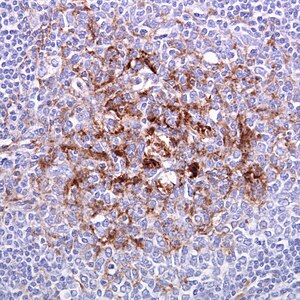 | |
| CJD患者的扁桃腺組織切片 | |
| 症状 | 失智症, 肌陣攣[*] |
| 类型 | 傳染性海綿狀腦病, disease of a particular individual[*] |
| 肇因 | 朊毒體 |
| 診斷方法 | 病史, neurological diagnostic techniques[*], 核磁共振成像, 腦電圖, 驗屍, 神經系統檢查[*], 組織病理學[*] |
| 治療 | 不明[*], 临终关怀 |
| 分类和外部资源 | |
| 醫學專科 | 神經學、傳染病學 |
| ICD-11 | 8E02.0 |
| ICD-10 | A81.0 , F02.1 |
| ICD-9-CM | 046.1 |
| OMIM | 123400 |
| DiseasesDB | 3166 |
| MedlinePlus | [1] |
| eMedicine | neuro/725 |
| Patient UK | [https://patient.info/doctor/Creutzfeldt-Jakob-Disease Creutzfeldt-Jakob-Disease 克雅二氏病] |
| MeSH | D007562 |
克罗伊茨费尔特-雅各布病(英語:Creutzfeldt-Jakob disease,縮寫:CJD),或稱克-雅氏症、克-雅氏病、克雅二氏症、克雅二氏病、庫雅氏症、庫賈氏症、克雅氏症、克雅氏病,是一種發生在人類身上的傳染性海綿狀腦病,分为普通和變种两個品種。其致病因子被認定為是朊毒體。目前是絕症,無法根治。
簡介
克雅二氏病(CJD)可按致病途徑分為偶發性(sCJD)、遺傳性(fCJD)、 醫源性(iCJD)(因醫療程序造成的感染)和變異型(vCJD)四類。偶發性克雅二氏病病因不明,它佔所有克雅二氏病患的百分之85至90,可能是美國用牛的血液和內臟餵豬造成帶有非典型狂牛症病原體的死豬(downed pigs)被人類食用而使得人類產生此種疾病;變異型克雅二氏病則是可傳染的,過去病例只在獵頭族发生。近年的變異型克雅二氏症可能透過食用或接觸患病動物,例如患有疯牛症的病牛或其他動物而感染。在美國紐約有患者吃過松鼠後染病的病例。
临床表现
病程可分为以下三个阶段:
- 进展期:主要为进行性的神经系统病情恶化,以小脑、锥体系和锥体外系的症状和体征为主。可表现为肢体僵直和震颤、感觉异常、共济失调、眼球震颤、语言障碍和失语等,并迅速进展为明显的精神衰退、半瘫、运动性失语,随之发生惊厥与昏迷,部分患者可出现大范围视觉异常,表现为视觉麻痹、变形和皮质盲。
- 终末期:患者进行性全身衰竭,最终往往死于肺炎或自主神经功能衰竭。CJD患者平均存活时间为6个月,约90%的患者发病后1年内死亡。
病理学
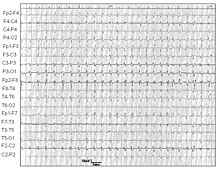
免疫组织化学和影像学表明,大部分CJD患者脑组织发生一定的萎缩,患者通常是脑室增大,而深灰色结构如尾状核、壳核和丘脑则发生萎缩,小脑也可由于缺乏灰质而可见薄层萎缩。与阿尔兹海默病不同的是,患者海马体不受影响。脑电图显示,绝大多数偶發性CJD患者在病程中可出现一种特异性的波形(周期性同步二或三相尖锐复合波),该波形特点为:严格的周期性脑电位,长度100-600毫秒,间歇500-2000毫秒;允许有泛平或侧向波形;要排除半周期性电活动,必须有连续5个间歇的长度差异小于500毫秒。其他类型CJD患者脑电图也有异常,但缺乏特异性。
诊断
WHO对于sCJD的诊断标准如下:
疑似病例诊断标准
以下临床特征中出现两项即可确定为疑似病例
- 进行性痴呆
- 肌阵挛,视觉或小脑性障碍,锥体束或锥体外系功能障碍,运动不能或缄默
- 病程中典型的脑电图改变,和(或)两年内死亡且CSF中14-3-3蛋白阳性
- 常规检查未提示其他诊断
确诊
除需符合上述四项标准外,还需具有以下神经病理学指标五项中的至少一项:
- 神经元丢失,胶质细胞增生,海绵状退行性变化,或脑组织免疫组化结果显示PrPSC阳性斑块
- 预先用蛋白激酶K处理后染色体PrPSC阳性
- 患者脑组织注射到实验动物后引起特征性神经退行性疾病
- 检测到PRNP基因突变的存在
流行病学

绝大部分CJD(不包括vCJD)为sCJD,约占85-95%,其余5-15%为fCJD,iCJD不足1%。世界范围内sCJD发病率为每年每1亿人中有1-10例病例,好发年龄集中于43-48岁,老年病例也可见报告。CJD男女比例约为1:1.2,无显著差异。
传播途径
此病的致病蛋白於一般消毒程序難以消滅,並可能透過接觸過患者體液、血液醫療和手術器具傳染他人,但日常社交接觸如握手、擁抱等則不會傳染此症。
限制捐血
为了保护血液供应通常會有以下措施:
以下情形通常會暫緩或禁止捐血行為:
- 1980-1996年間(狂牛症大流行期間)曾至英國旅遊或居留時間合計超過短時間者;
- 1980年以後曾在英國接受輸血或使用製劑(有些擴大到法國);
- 1980年以後曾於歐洲旅遊或居留時間合計超過長時間者。
以下情形通常會直接禁止捐血行為:
- 曾注射人類腦下垂體生長荷爾蒙者
- 曾注射人類腦下垂體親生殖腺素(human pituitary gonadotropins)者
- 曾注射牛胰島素等生物製劑者(尤其是英國牛源製造之胰島素者)
- 曾接受硬腦膜移植者
- 家族中有克雅二氏病患者(尤其是二等親內血親中)
治疗
包括CJD,所有朊粒病均缺乏特效药物治疗,主要为支持治疗。有关金刚烷胺、阿糖腺苷等可以稳定或改善病情的个别报道尚待进一步证实。另有研究表明阿昔洛韦、干扰素和两性霉素B对朊粒病无效。自2003年起,有一種新的藥物Pentosan Polysulphate(簡稱PPS )開始被實驗性地使用在一位患者Jonathan Simms 身上,該患者一度情況穩定,但是死於2011年。然而,PPS至多只能達到抑制病情的效果,病症仍沒有辦法根治。而且英國衛生部門的克雅氏病治療諮詢小組(CJD Therapy Advisory Group to the UK Health Departments)指出該資料並不足以支持PPS 是一個有效治療的聲明,並且建議更多動物樣本的研究才是適當的。
一個2007年對於26個使用PPS 治療病人的報告也因為缺乏可以接受的客觀因素而沒有發現其為有效的證明。
類似病症
相似症狀的庫魯病,最早在1950年發現於新幾內亞的部落,該部落有攝食死亡親戚的腦部和其他組織的習慣。其症狀為患者的腦部組織會產生空洞、海綿化,並導致腦部退化、肌肉失去協調和痴呆,然後死亡。在澳洲政府禁止食人行為後,庫魯病也就消失了。
克雅二氏病的症狀和庫魯病相似,但克雅二氏病為全球性的疾病,約有一百萬分之一的人會得此病,通常發生在50歲以上的人。
1913年,兩名德國醫生,庫茲菲德(Hans Gerhard Creutzfeldt)和雅各(Alfons Maria Jakob),在大約同時第一次記錄了這種新病,因此此病以他們兩人的名字來命名。
1982年,加州大學舊金山分校的史坦利·布魯希納發現了這一類疾病的病原體朊毒體蛋白,為此他得到了1997年的諾貝爾生理醫學獎。
近年的新變种——變異型克-雅二氏病,可能透過食用或接觸患有疯牛症的病牛而感染。
参见
參考文獻
外部連結
- 勞永樂專科醫生. 克雅二氏症與牛肉解禁. 《醫·藥·人》. [2012-05-10]. (原始内容存档于2020-12-12).
- 克雅二氏症 (页面存档备份,存于互联网档案馆)
| |||||||
| ||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||