
| Myasthenia gravis | |
|---|---|
 | |
| Detailed view of a neuromuscular junction: 1. Presynaptic terminal 2. 肌膜 3. 突触小泡 4. 菸鹼型乙醯膽鹼受體 5. 線粒體 | |
| 类型 | 神经肌肉传递障碍[*], autoimmune disease of musculoskeletal system[*], immune-mediated acquired neuromuscular junction disease[*], 免疫系统疾病[*], autoimmune disease of peripheral nervous system[*], disease of a particular individual[*] |
| 分类和外部资源 | |
| 醫學專科 | 神經學 |
| ICD-11 | 8C60 |
| ICD-10 | G70.0 |
| ICD-9-CM | 358.0 |
| OMIM | 254200 |
| DiseasesDB | 8460 |
| MedlinePlus | 000712 |
| eMedicine | neuro/232 emerg/325 (emergency), med/3260 (pregnancy), oph/263 (eye) |
| Patient UK | [https://patient.info/doctor/myasthenia-gravis-pro myasthenia-gravis-pro 重症肌無力] |
| MeSH | D009157 |
| Orphanet | 589 |
重症肌無力(英語:myasthenia gravis,簡寫MG)是慢性神经肌肉传递障碍的自身免疫性疾病,會造成不同程度的肌肉無力。最常影響眼部、臉部及吞嚥相關的肌肉。會造成复视、眼睑下垂、說話困難及行走困難等症狀。重症肌無力可能會突然發生。患者多半有胸腺過大或是胸腺瘤的情形。
重症肌無力屬於自體免疫性疾病,是因為抗体堵塞或是破壞神经肌肉接点中的尼古丁受器,因此神經脈衝無法引發肌肉收缩。有一種罕見的神经肌肉接点遺傳疾病也會有類似症狀,稱為先天性肌无力综合征。肌無力的母親所生的嬰兒,在出生後頭幾個月也有會有類似症狀,稱為新生兒肌無力。診斷可以透過檢查特殊抗體的血液檢查、騰喜龍試驗或是神經傳導研究進行。
重症肌無力的治療方式多半是透過像新斯的明或吡斯的明等乙醯膽鹼酯酶抑制劑來治療,有時也會使用强的松或是硫唑嘌呤等免疫抑制藥物。胸腺切除術可能可以改善一些病患的症狀。若是突發性的病情,可能會使用血漿分離術及高劑量静脉注射免疫球蛋白治療。若呼吸相關肌肉非常無力,需要用呼吸器幫助呼吸。
每百萬人中,約會有50至200人罹患重症肌無力,每年每百萬人中的新診斷病例為3至30人。因為相關疾病意識的提高,也較容易診斷到相關病例。最常見的是四十歲以下的女性以及六十歲以上的男性,孩童的病例並不常見。大部份的病患在治療後可以有較正常的生活,其预期寿命也和一般人相當。重症肌無力的英文Myasthenia gravis原自希臘文的mys(肌肉)及astheneia(虛弱)以及拉丁文的gravis(嚴重)。
症狀
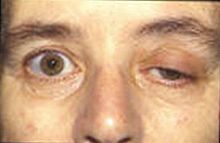
重症肌無力的徵狀是容易疲勞,肌肉會在活動期間逐漸變得軟弱,經過一段時間的休息便能恢復。肌肉能夠控制眼皮及眼球的活動,及面部表情、脖子和肢體運動,咀嚼及吞嚥、清楚的發音/談話及控制呼吸的肌肉都是特別容易受影響。定期的身體檢查能保持身體於正常的狀態。
人類的免疫系統的紊亂起源可能是突然的,但紊亂的症狀通常是斷斷續續的。由於徵狀的微妙或易變,會引致延遲診斷。 在許多個案中,首先患者注意到的徵狀是眼皮肌肉容易疲勞和軟弱。另外,吞嚥困難和言語不清也可能是最初徵兆。在不同的病人中肌肉無力的程度有著很大的差異,範圍能從局部的形式縮窄至眼部的肌肉(視覺的重症肌無力),到局部嚴重徵狀或許多肌肉 - 有時包括控制呼吸的肌肉也會受到影響。這些症狀的類型和嚴重性變化,可能包括不對稱的眼皮下垂(一隻或雙眼的眼皮下垂)、複視(雙眼看到重影)。由於肌肉無力控制眼球移動,頸部、手臂、手指、手部、腿部的關係,會引致呼吸短暫而急促、斷續的講話、吞嚥困難、在面部表情上只有一個變化、說話經常出現鼻音不穩定或步履不穩的步態。
預後
經過治療,預期患者會有正常的生活,除非患者有惡性腫瘤 (預期的壽命是根據腫瘤本身而非重症肌無力)。生命的質素是根據起因與嚴重的變化。用於控制重症肌無力的藥物效力會隨時間而減弱,或是會產生嚴重的副作用,例如免疫力下降。有大約10%的重症肌無力病人會有胸腺腫瘤,胸腺切除手術對這些病人非常有效,症狀或會長期消失。但是,大部份的病人需要一生接受治療,而治療效果則因人而異。
重症肌無力並不是一個漸進性的病,不會隨著年齡增長而變差,但症狀會不定時出現。有一些病人的症狀會在3至5年後減輕。
流行病學
重症肌無力會在不同的種族、性別中發生,它通常會發生在40歲以下的女性和50-70歲的老年人身上,但其實所有年齡階層的人都可能患上此病。年輕的病人通常不會有胸腺瘤。 在美國,每100,000人中就有20個人罹患此症,年齡介乎20至40歲、有家族病史、服食「滿克特 (Metalcaptase)」(D-penicillamine),或者患有其他自體免疫病症的人會有較高的機會率患上重症肌無力。
分類
重症肌無力廣泛被接受的分類是以美國臨床分類為基礎:
- 第一類:出現眼肌軟弱或疲勞情況,眼皮可能下垂(ptosis),並沒其他證據顯示身體其他地方出現肌肉軟弱或疲勞。
- 第二類:眼肌軟弱或疲勞情況較嚴重,其他肌肉軟弱或疲勞情況溫和。
- 第2A類:主要在肢體或軸向肌肉。
- 第2B類:主要在延髓(bulbar)及/或 呼吸肌肉。
- 第三類:眼肌軟弱或疲勞情況嚴重及其他肌肉出現軟弱情況。
- 第3A類:主要在肢體或軸向肌肉。
- 第3B類:主要在延髓(bulbar)及/或 呼吸肌肉。
- 第四類:眼肌軟弱或疲勞情況嚴重及其他肌肉軟弱情況嚴重。
- 第4A類:主要在肢體或軸向肌肉。
- 第4B類:主要在延髓(bulbar)及/或 呼吸肌肉。(需要以胃管進食但不需要插管)
- 第五類:需要插管以維持呼吸。
病生理學
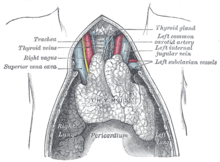
重症肌無力是一個发生于肌肉突触的自体免疫疫疾病。当人体的免疫系统出现紊乱,并且产生会攻击自己身体组织的有害抗体时,这种病症就会发生。这种有害抗体进攻人体的一种正常蛋白,这种蛋白通常为乙酰胆碱受体,或者MUSK受体。 另外还有一些不常见的有害抗体包括LRP4, Agrin等。 當各種相似的疾病與其他疾病交叉感染的媒體,這是沒有已知的起因(病原體)令人患上重症肌無力。有一個微細起源的傾向:特別的HLA類型似乎為MG預先處理 (DR1的B8及DR3為特定的視覺上的重症肌無力)。有75%患者的胸腺有異常;其餘25%有腫瘤(良性或惡性),及經常會找到其他異常。此症通常會在切除胸腺後保持穩定。

在重症肌無力,抗體通常會直接共同對抗菸鹼酸的乙醯膽碱感受體(nAChR),那些運動終板的感受體刺激神經傳送素乙醯膽素來刺激肌肉的收縮。抗體以某種形式削弱乙醯膽碱的能力來約束感受體。其他導致感受體的破壞,由血清中的補體固定方法或誘導肌肉細胞通過胞吞作用去消滅感受體。
抗體是由血漿細胞產生,從B細胞取得。辅助性T细胞刺激使B細胞轉變成浆细胞。為了這個激活過程,輔助性T細胞必須首先被自己的細胞活化,那是受到T細胞感受體(TCR) 的約束,到乙醯膽素感受體抗原性縮氨酸碎片(epitope) resting within the MHC of an 抗原代表細胞。當胸腺在T細胞的生長擔當一個重要的角色,和TCR 重症肌無力的篩選是與胸腺瘤有密切關係。然而這精確的機制不能有說服力地澄清,儘管在胸腺切除術施行在沒有胸腺瘤的重症肌無力患者身上有正面效果的說法。
在正常肌肉收縮,nAChR累積的活化作用導致鈉的積聚,這引致鈉化離子的匯集,那就反過來導致肌细胞和肌肉細胞退化。這離子的匯集通過T細管落到細胞膜,及肌漿網(sarcoplasmic reticulum)通過鈣的複合物導致鈣的釋放。只有當鈣於肌肉細胞的水平高得足夠令它收縮。nAChR的功能數目減少因此使退極化削弱了肌肉收縮。事實上,重症肌無力導致神經元動作電位到肌肉抽搐率的變化,從非病理學的一個到一個的比率的變化。
最近認識到第二類別的重症肌無力是由於自身的抗體對抗MuSK蛋白質 (肌肉獨有的激酶),酪氨酸激酶感受體 (tyrosine kinase receptor),那是構成神經肌接合點的必需物。通常抗體對抗MuSK以禁止MuSK的信號被它的神經衍生配體 (nerve-derived ligand)誘導,聚集素。結果是下降神經肌接合點的顯著性,及重症肌無力隨後的症狀。
人類服用青黴素能顯露重症肌無力的症狀。他們的抗體滴定濃度通常與重症肌無力相似,但當藥物用法中斷了,症狀及滴定濃度就會消失。
重症肌無力與其他自我免疫的疾病是相類似,有5%的個案是有遺傳的傾向。這是與某些基因的變化如 HLA-B8及DR3的頻率上升有關聯。重症肌無力患者比一般人口患上自我免疫的疾病有更高的頻率。特別要提及的是現存的甲狀腺疾病中的甲狀腺官能不足病會促使嚴重的惡化。
Rapsyn蛋白質會使乙醯膽碱感受體聚集成群及固定,研究最終會帶出新的治療方案。
診斷
重症肌無力是很難診斷的,因為症狀是難以捉摸,和較難分別出是正常變形與其他神經紊亂。一個全面的身體檢查能顯露出病人容易疲累的症狀,當經過休息之後能改善疲累的情況,及重複和延長測試會令疲累的情況惡化。使用冰到弱的肌肉部分能有代表性地改善那部分的肌肉強度。以下所提及額外的測試會經常執行。此外,在療程中的良好反應都會被考慮為病理學中自動免疫的標記。
臨床檢查
很多肌肉都能測試出肌肉疲累。一個徹底的檢查包括:
- 病人的眼睛向上望和向側看30秒:眼皮下垂(ptosis)及複視(diplopia)。
- 橫臥時望向腳60秒。
- 持續把手臂伸直60秒。
- 膝彎約10次。
- 腳尖跟腳跟步行30步。
- 仰臥起坐5次,需要完全躺下及坐起來。
- "窺視標誌(Peek sign)":完成眼瞼邊緣最初的並列後,病人很快(30秒內)開始分開和眼球的鞏膜開始表現出來。
血液測試
如診斷是有懷疑的,血清學 (serology)會用於血液測試來鑑別某抗體:
- 有一個測試是為抗體阻擋乙醯膽碱感受體的。這個是合理的敏感性測試。這測試的合理敏感性是 80–96%,但重症肌無力限制了眼球肌肉 (視覺的重症肌無力), 這個測試或許是消極至個案的50%。
- 病人體內沒有抗體的比例以乙醯膽碱感受體的抗體抑制MuSK蛋白質。
- 在特殊的情況(減少反映促進增加、現行共同的自律的特色、懷疑腫瘍或癌的出現,特別是肺部,反覆的EMG測試能增加或促進其存在性)。進行肌肉萎縮症候群(Lambert-Eaton syndrome)測試,並從中找到其他的抗體。
短效膽素酯酶抑制劑(Edrophonium)的測試
短效的膽碱酯酶抑制劑 - Edrophonium的測試是很少用以確認重症肌無力的存在。當其他調查不能確定的診斷時,情況會限制了它的應用情況。 這個測試要求短效的膽碱酯酶抑制劑 - Edrophonium氯化物於靜脈內進行管理或使用新施得明藥物能夠產生膽素酯酶及暫時性增加乙醯膽碱在神經肌肉接頭的水平,從而阻礙乙醯膽碱的分解。有眼部肌肉疲弱的重症肌無力患者,短效的膽碱酯脢抑制劑 - Edrophonium氯化物能在短時間內減輕軟弱的情況。
掃描

胸腔的X光掃描是經常會做的,它可能指出其他的診斷結果(例如肌肉萎縮症引致的肺部腫瘤)。這也許能辨認出橫膈膜的腫瘤,但電腦掃描 (CT) 或磁力共震掃描 (MRI) 都是比較準確去辨認腫瘤的存在,及通常都是這樣做。
肺功能測試
關於病人有否足夠的呼吸能力,肺功能測試(Spirometry) 可用以評估其呼吸機能。不自然維持生命能力(vital capacity)的間隔監測是為了不會錯過肌肉逐步惡化的過程。嚴重的重症肌無力可能會使呼吸肌肉精疲力竭,而導致呼吸衰竭。
神經生理學
重症肌無力患者的肌肉力量很容易疲勞,通常在臉部做電刺激測試,在重複的刺激時,他們的反應會不及健康的個體。當利用電力脈衝重複刺激肌肉時,肌肉的疲勞程度可被量度出來。這稱為「反覆神經刺激測試」。在單一力量 - 肌動電流描記術 (electromyography)之中,這是認為重症肌無力最敏感的測試(雖然不是最具體的)。肌動電流描記術是利用一根幼針插入肌肉內以電位來記錄個別肌肉力量。由此得出兩組肌肉力量屬於相同的motor unit及在燃燒的圖案來量度短暫的變化的傾向,由此得出診斷結果。
病理學研究
如診斷及患者的肌肉狀況是有所懷疑的話,肌肉活組織切片檢查法有需要進行。螢光染色的抗體顯示IgG抗體在神經肌接合點上。(注意這不是導致重症肌無力的抗體發螢光,但是次等的抗體會直接產生對抗。)肌肉在電子顯微鏡下顯示感受體摺疊及失去摺疊的傾斜,與加寬了的突觸裂縫連在一起。這些技術目前用於研究而非用於診斷上。
治療
治療的方法是透過藥物及/或進行手術。藥物治療主要由膽素分解酶抑制劑構成,能直接改善肌肉功能,和抑制免疫力的藥物能夠節省自我免疫的過程。Thymectomy 是一種外科手術用的方法來治療重症肌無力。對於緊急的治療,血漿置換術 (plasmapheresis) 或靜脈內的免疫血球蛋白可用以暫時性的方法從血液循環中去清除抗體。
藥物
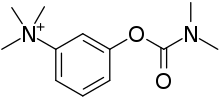
- 膽碱分解酶抑制劑:新施得明及 Mestinon (美定隆,俗稱大力丸) 能透過減慢自身一種稱為膽碱分解酶(它會分解在運動終板的乙醯膽素)的酵素作用,因此可延長神經遞質刺激感受體的時間以改善肌肉功能。通常醫生會處方從少劑量開始,如3x20mg的pyridostigmine,逐步增加直至達到預期效果。如在餐前30分鐘服藥,進餐時的症狀會較溫和。至於副作用,如汗水較多及腹瀉,而腹瀉可以通過增加顛茄鹼(atropine)來抵抗。Pyridostigmine 是短暫的藥物,其半衰期約4小時。
- 抑制免疫力的藥劑:Prednisone (強的松,也稱波尼松,一種腎上腺皮質類固醇)、Cyclosporine (环孢素,拒絕反應抑止藥)、Mycophenolate Mofetil 及 Azathioprine可能會用上。患者通常會把這些藥物與膽碱酯脢抗化劑結合使用來治療。一些抑制免疫力藥劑的治療需要服用至效果被注意到的幾星期至幾個月後。其他免疫控制物質,如:預防免疫系統調查以調節乙醯膽素感受體的藥物。
- 中醫藥方治療:重症肌無力屬於中醫範疇的痿症,根據中醫學說,脾臟主四肢主肌肉,所以重症肌無力是脾陽的問題,近年來以補中益氣湯水為治療基礎取得很好療效,而且没有激素治療的後遺症。不過中醫藥方案主要是起到輔助和調理的作用。
血漿置換術及免疫血球蛋白
如重症肌無力是嚴重的話,如重症肌無力的病情危險期,血漿置換術是可用以從循環系統去除推定的抗體。而且,靜脈內的免疫血球素可用以約束循環的抗體。以上的治療相對地有短暫的好處,典型地以星期計算。
手術
胸腺切除術 (thymectomy),是以外科手術移除胸腺,當有胸腺瘤是必要做的,這有鑑於潛在的瘤的產生,亦即是腫瘤。但是,當患者不提供胸腺異常的情況,這程序是較有爭議的。儘管那些患者以胸腺切除術改善了病情,有些患者會遇到嚴重的exacerbations及富爭議的概念 - "therapeutic thymectomy",患者會有胸腺增殖(thymus hyperplasia),這是經過許多專家的討論及努力之中毫不含糊的回應這重要的問題。
這裡有些外科的方法來移除胸腺瘤:通過胸骨 (transsternal), 通過頸部(transcervical)及通過胸廓 (transthoracic)。通過胸骨(transsternal)的方法是最常用和通過胸骨使用同一縱長的切口,那是用於開心手術的。通過頸部(transcervical)的方法較少侵略性的程序,這容許通過頸部細小切口來移除整個胸腺。這個方法與通過胸骨的方法沒有分別在改善症狀和較通過頸部的方法較少侵略性。
然而有胸腺瘤的患者,最重要是所有胸腺瘤組織已被去除,胸腺能重新繼續生長。胸腺瘤可能是惡性的和被認為是其他疾病的開始。正因如此,很多外科醫生只會建議全面的sternotomy方法予胸腺切除術。
胸腺瘤是相對地罕見於較年輕(即40歲以下)的患者,但似非而是地尤其有全身性的重症肌無力的年輕患者而沒有胸腺瘤得益於胸腺切除術。當然,切除術也顯示那些患者有胸腺瘤,但對重症肌無力的症狀改善則可能性很少。
兒童
兒童患上的重症肌無力可分為﹕
- 嬰兒期的重症肌無力﹕有12%患有重症肌無力的懷孕婦女會將致病的抗體經胎盤傳至胎兒體內,令嬰兒出生之後出現嬰兒期的重症肌無力,症狀在會出生後的兩天內出現,數星期之後便會消失。患有重症肌無力的孕婦有時候症狀會在懷孕期間減輕,但在生產之後可能會轉差。
- 先天性重症肌無力﹕即使母親沒有患上重症肌無力,嬰兒亦有可能患有重症肌無力,但這非常罕見,這稱為先天性重症肌無力。此不同於一般的重症肌無力,它不是一種自體免疫力疾病,它的成因是由基因突變而形成的神經突觸畸形,所以先天性重症肌無力是一種罕見的遺傳病,已知的相關基因突變大約有11種,而它們的遺傳方式都是隱性遺傳。
- 幼年的重症肌無力﹕在孩童時期患上的重症肌無力,但不屬於上述兩種。
先天性重症肌無力會導致肌肉無力及疲勞,症狀與重症肌無力相似,症狀通常在兩歲前出現,但有幾種先天性重症肌無力會在70歲時才發病,先天性重症肌無力的診斷決定於﹕
- 症狀在嬰兒時期或孩童時期發生
- 肌肉無力的情況在會肌肉疲倦時加劇
- 在低頻的肌電圖檢查中複合肌肉動作電位波(compound muscle action potential)反應下降
- 沒有anti-AChR或MuSK抗體
- 對抑壓免疫力的治療沒有反應
- 有類似先天性重症肌無力的家族病史
先天性重症肌無力可以由輕微到嚴重,即使不同的病人擁有同一樣的突變基因,他們的症狀嚴重程度也會有所不同。大部份的先天性重症肌無力的病情也不會轉差,在某幾種的先天性重症肌無力中,症狀更可能隨年齡而減弱,會隨時間而愈趨嚴重的先天性重症肌無力非常罕見。
懷孕
長遠而言,懷孕不會對重症肌無力有影響。有10%的初生嬰兒及父母的情況是出生與短暫的(周期性)初生的重症肌無力通常會引起飼育和呼吸困難的影響。初生的重症肌無力一般會以弱於飼育及通常會低肌肉張力。其他轉述的症狀包括抽搐地哭,面部兩側麻痺(身體某部分麻痺或癱瘓)或輕度癱瘓及輕微的呼吸困難。有初生重症肌無力的兒童能對乙醯膽素酯脢抗化劑有很好的反應。有1/3的母親患上加劇的重症肌無力和令病情惡化,這通常發生於懷孕的第一期。於懷孕的第二期及第三期,母親的症狀會有改善,有些母親更能夠完全的減輕病情。抑制免疫力的治療法應在懷孕中被維護,這樣能夠減少初生的肌肉軟弱的機會,也能控制母親的重症肌無力。
很少時候,初生嬰兒出生時有先天性多發性關節彎曲,第二是子宮深處軟弱。這是由於母親的抗體的目標是初生嬰兒的乙醯膽素感受體。有些例子,母親會是無症狀的 (asymptomatic)。
歷史
- Mary Broadfoot Walker(1888-1974)是首位發現有效的乙醯膽胺酯化醊抑制劑(physostigmine)來治療重症肌無力的英國內科醫生。
外部連結
- The Myasthenia Gravis Foundation of America (页面存档备份,存于互联网档案馆)
- The Myasthenia Gravis Association (MGA) in the United Kingdom & the Republic of Ireland
- The Myasthenia Gravis Coalition of Canada (页面存档备份,存于互联网档案馆)
- The Hong Kong Association of Myasthenia Gravis 香港肌無力協會 (页面存档备份,存于互联网档案馆)
- 台灣肌無力症關懷協會
參考資料
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||