
| 進行性骨化性纖維發育不良 | |
|---|---|
| 同义词 | 石人綜合症、珊瑚人、石頭人 |
 | |
| 進行性骨化性纖維發育不良,導致結締組織受傷後再生成骨骼 | |
| 类型 | 結締組織病, 遺傳性疾病, primary bone dysplasia with progressive ossification of skin, skeletal muscle, fascia, tendons and ligaments[*], autosomal dominant disease[*], disease of a particular individual[*] |
| 分类和外部资源 | |
| 醫學專科 | 遺傳性疾病 |
| ICD-11 | FB31.1 |
| ICD-10 | M61.10、M61.111、M61.112、
M61.119、M61.121、M61.122、 M61.129、M61.131、M61.132、 M61.139、M61.141、M61.142、 M61.143、M61.144、M61.145、 M61.146、M61.151、M61.152、 M61.159、M61.161、M61.162、 M61.169、M61.171、M61.172、 M61.173、M61.174、M61.175、 M61.176、M61.177、M61.178、 M61.179、M61.18、M61.19 |
| ICD-9-CM | 728.11 |
| OMIM | 135100 |
| DiseasesDB | [1] |
| eMedicine | 1112501 |
| Orphanet | 337 |
進行性骨化性纖維發育不良(英語:Fibrodysplasia Ossificans Progressiva,簡稱FOP)又稱進行性骨化性纖維增殖症或是進行性骨化性纖維炎,舊稱為進行性骨化性肌炎(英語:Myositis Ossificans Progressiva,簡稱MOP)或稱進行性肌肉骨化症,此疾病不應與骨化性肌炎搞混,因後者通常是創傷後所發生,且此疾病不只發生於肌肉,也發生於其他纖維組織。
FOP是一種極其罕見的遺傳性疾病,最早的案例發生於1692年,之後325年到2017年為止,全世界的病例僅700餘件,因為病例過少,正確的發生機率無法統計,被認為是世界上最罕見的疾病之一。
FOP的發生大多源自於突變,使得身體修復系統異常,患者身體的肌肉以及結締組織會發生異位骨化,尤其在受傷後,受傷處的骨化會加重,使得病症更為嚴重。
流行病學
FOP為全世界最罕見的疾病之一,發病率尚未有定論,普遍認為是0.61/百萬人左右。大多數報導的病例均發生在白種人中,但所有種族均有可能發生,且男女發病比為4:1。
巴西FOP協會截至2005年,登記了49個病例。國際上則有IFOPA(International FOP Association,國際FOP協會)提供關於FOP流行病學的相關資料,以及為全球大部分確診患者提供協助。
病因
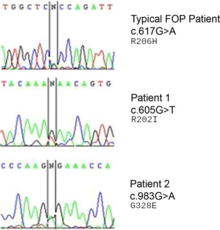
該疾病源於人第2對染色體2q24.1區域的ACVR1基因座位上的完全顯性等位基因發生的雜合突變。其所影響的BMP1活動異常。由於過度活化的關係,結締組織及肌肉組織會轉變成次級骨骼,使內皮組織轉變為間充質幹細胞,最後形成骨骼。
另外BMP4的異常表現也被認為是導致了異位骨化的致病因子之一。BMP4並不直接變異,而是與BMP4相關的訊號傳導因子突變,而Noggin蛋白是BMP4的拮抗因子(抑制BMP4),患者身上可查到Noggin蛋白基因的突變,此突變位於17q22。此外,有時在42bp處也能發現Noggin蛋白基因的缺失,可認為皆為FOP的致病原因之一。
大多數的病患是由於配子中發生自發突變,因為其遺傳性,絕多數的FOP患者不能或是不願選擇生小孩。也有類似但傷害性較低的疾病纖維發育不良,則由雜合突變所產生。
執行骨化的基因通常於胎兒形成骨骼後便關閉,但FOP患者中的基因則持續發揮作用。患者在結締組織和肌肉細胞受損或異常傳遞凋亡訊息時產生骨骼修復酶(Enzyme for bone repair),這導致淋巴系統存在過量的BMP4並在免疫系統作用時產生效用。產生的新骨獨立於正常骨骼,形成許多離散骨架,不過這些骨骼仍可以與正常骨架結合。
歷史
FOP最早的案例是在1692年由蓋‧帕丁(Guy Patin)在兒童身上發現,以及弗里克(Freke)在1739年對此病症做了描述,1918年,朱麗葉斯‧羅森斯吞(Julius Rosenstirn)所著的書籍中詳細記錄了115件FOP患者。在當時,此疾病被命名為進行性骨化性肌炎(Myositis Ossificans Progressiva),代表肌肉逐漸骨化的現象。但由於此疾病亦發生於關節囊、韌帶等部分,維克托·麥庫西克(Victor Almon McKusick)在1970年代,將其更名為進行性骨化性纖維發育不良(Fibrodysplasia Ossificans Progressiva)。
世上最著名的FOP案例為哈里·雷蒙·伊斯特拉克(Harry Raymond Eastlack),FOP在他十歲時發病,在1973年11月即將40歲時死於肺炎,他的身體嚴重骨化,只剩下嘴巴可以動。死後,他的遺體被捐給科學研究,其骨架現展示於費城馬特博物館。
症狀
FOP最顯著的臨床特徵為先天性腳大拇趾畸形,大約有75%~90%的病例在出生時即可觀察到此特徵,也約有一半的病例在出生時可觀察到大拇指畸形。此外,兒童期開始之後的外骨骼形成也是典型特徵之一。
早期的發病,會出現局部紅腫疼痛,數日至數周後消退,接著會產生局部僵硬感。因為軟組織內可能產生硬骨顆粒,可能刺激組織產生痙攣。 一段時間後,疾病可能擴散至全身,尤其好發於頸、肩、胸、腰或是其他關節處。發病後大約10年,患者的脊椎以及肩會受到嚴重的限制;大約20年後,髖關節也會失去移動能力;多數的患者在發病後30年只能坐在輪椅上。
FOP除了自發性的惡化之外,也可能透過肌肉注射、疫苗、局部麻醉、活體組織切片、靜脈注射或是任何外傷而惡化。因此應避免任何鈣化結節的切片檢查,可能造成再次骨化,甚至比原先更為嚴重。
診斷
由於外觀體徵明顯,可由觀察外表或著透過X光進行診斷。但新生兒身上廣泛的特徵──腳趾畸形,可能是較為常見的先天性外翻趾(Isolated congenital Hallux Valgus)而非FOP所造成。 由於任何創傷都可能造成惡化,最好在新生兒時期便進行診斷,以避免可能發生的傷害。透過無創檢察來早期診斷及監測疾病發展,骨顯像用於顯示異位骨化,而斷層掃描及核磁共振則可用來檢測不明顯的新病灶。
適合用來測定骨分化的人類乳牙幹細胞(SHED)顯示FOP患者身上的SHED細胞有更高水平的鹼性磷酸酶以及Runx2 mRNA濃度。因此在臨床上可透過檢測這些物質水平來診斷。
治療
目前並無有效的治療方式,有一部分文獻表明羟基乙叉二膦酸(Etidronate Ethane –1-Hydroxy-1-diphosphonate)對部分患者的急性期有效,但仍在研究階段,且仍未被證明有效。由於BMP受體的活化突變揭露了潛在的關鍵因子,可能可以作為重要的藥物靶點,為醫學研究的重要目標。而確診FOP之後,應盡量避免任何傷害及侵入性手術,包括抽血。
實驗及研究
實驗中,FOP在急性期,對於鈣、磷以及副甲狀腺素的代謝正常。除此之外,對於骨礦物質的代謝,透過生化分析亦未發現異常。疾病爆發期間,尿液中可發現鹼性成纖維細胞生長因子(bFGF)水平的升高。
由於FOP相當罕見,很大程度的阻礙了人類對FOP的研究。不過Rothschild等人在東南亞的小鼷鹿身上發現該物種的成年雄性──無論飼養或是野生──身上,有自發性的FOP產生,可能提供了科學家的研究機會。
2015年8月,美國FDA授予拉荷亞(La Jolla)製藥公司針對FOP的兩種化合物孤兒藥的地位,藥內為小分子的蛋白激酶抑制劑,目的在阻斷ACVR1。
同時,加拿大Clementia Pharmaceuticals公司開始招募6歲以上的患者,參與其藥物Palovarotene的二期實驗。臨床研究顯示視黃酸以及維甲酸受體γ激動劑,可透過抑制BMP途徑中的第二信使系統,阻止異位骨化的形成。
參見
註解
參考來源
外部連結
| |||||||
| ||||||||||||||||||||||
|